

It has been nearly a decade since whole genome sequencing was first used clinically to diagnose a child with a rare idiopathic disease.1 Since then, thousands of rare disease patients have benefited from advances in next-generation sequencing (NGS) methods. Sequence alignment and variant calling tools have improved substantially, and reference databases have expanded in scope and depth. While these advances have improved the accuracy of sequence data, the most significant gains in clinical applications of sequencing have come from the development of advanced analytical methods.
This white paper outlines the evolution in genome data analytics from simple filtering approaches to more sophisticated AI algorithms for identifying the causal variants in rare disease patients. For over a decade, Fabric Genomics has been a leader in developing cutting edge AI approaches for analyzing genomes. Here we describe how Fabric’s successive algorithms, VAAST, PHEVOR, and now GEM, have continually pushed the boundaries to accelerate the identification of rare disease variants in patients.
Rare Genetic Diseases
There are approximately 7000 rare diseases with either known or suspected genetic etiology that have been described. Most are exceedingly rare, affecting fewer than <1/2000 to <1/10,000 individuals. Collectively, they impact about 6% of the population. Most rare diseases present in childhood, and about 80% are thought to be genetic or have a genetic component. Some present immediately at birth, requiring hospitalization and intensive care, while others appear several years later in childhood or even adulthood and can go unrecognized and undiagnosed.
Undiagnosed genetic diseases can take a physical, financial, and emotional toll, not only on the patient but their families and healthcare institutions as well. Sequencing presents a viable testing option for these patients. Clinical whole exome (WES) or whole genome (WGS) sequencing results in a diagnosis in an average of 36% (WES) or 41% (WGS) of cases, although this number can vary by condition type, age of onset, and prior diagnostic testing.2
Sequencing Methods and Diagnostic Approaches
Many of the clinical genetic tests available today are based on NGS. These tests range from the analysis of single genes or gene panels to a more comprehensive analysis of a patient’s whole exome or whole genome. WES queries the protein-coding regions of nearly all of the ~20,000 genes in the nuclear genome. As most Mendelian disease-causing variants are located in the exome, it represents an efficient approach for searching for the genetic cause of a rare undiagnosed disorder.
WGS, in contrast, queries not just the exome but the other >98% of the genome that does not code for proteins as well. WGS is several times more expensive than WES but has several advantages. Compared to WES, WGS offers better coverage across the genome, resulting in fewer gaps where variants might be missed. WGS includes the detection of variants in the mitochondrial genome as well. WGS can detect not only the variants in the exome but also those intergenic and intronic pathogenic variants, which are an infrequent yet increasingly recognized source of disease-causing variants. WGS is also capable of detecting in a single assay not just single nucleotide variants (SNVs), but large structural variants (SVs) and copy number variants (CNVs) of varying sizes, which can capture an additional ~10% of individuals with a clinically significant result.3
Clinical analysis of a rare disease patient usually begins with a detailed clinical assessment. For patients with a well-defined disorder, limited testing with a single gene or gene panel may be sufficient. For patients whose condition is not well-defined or who fail to find a causal variant using gene panel testing, WGS or WES may be appropriate. The choice of whether to undergo WGS or WES is mostly an economical one. Still, mounting evidence suggests that WGS/WES is cost-effective when used as a first-line genetic test, especially early in a diagnostic journey4-7. Analysis typically involves sequencing the genome of an affected child and sometimes other affected and/or unaffected nuclear family members (i.e., parents or siblings) if available.
Genome Analysis – Stages
Genome data analysis is divided into stages.

- Primary analysis involves capturing, and translating into digital format, the raw data that comes from a sequencing machine. This process is carried out directly by the sequencing instrument.
- Secondary analysis involves aligning the sequence reads to the reference genome and calling variants, trying to discard artifacts and sequencing errors.
- Tertiary analysis includes annotating variants with information from various databases and interpreting that data using manual or automated methods.
Laboratories conducting genome analysis can choose to build their own custom bioinformatics pipeline for secondary and tertiary data analysis, piecing together freely available tools developed in academic or government labs. Increasingly, labs are turning to packaged software solutions that provide reliable, turn-key alternatives for consistent, high-quality data interpretation. Labs can thus avoid the hassle of continual maintenance and updates needed to keep pace with this rapidly evolving field, allowing them to focus resources on their areas of expertise. Fabric Genomics offers a comprehensive suite of tools, including sophisticated AI algorithms and data sources aligned with best practices, all in an intuitive workspace that can be accessed through a secure web interface.
Genome Analysis – Data Used in Analysis
The goal of clinical genome analysis is to query the estimated four to five million variants in a patient’s genome to find the one or two variants causing the disease. The basic assumptions underlying most genome analysis approaches are: 1) That the disease-causing variant(s) most likely disrupts a gene in such a way as to damage its protein product; 2) That the disease-causing variant(s) is rare; and 3) That the gene is inherited in a Mendelian fashion, implying that other affected family members should also have the causal variant(s) and that parents contribute variants to the patient that, in combination, cause disease.
Identifying protein-altering variants
Information about whether the variant is protein-altering comes primarily from the tertiary analysis pipeline. For a given gene transcript, it determines whether the variant is in the coding region, a splice region, or elsewhere. For coding region variants, it determines whether the change results in a synonymous (no change in amino acid) or non-synonymous amino acid substitution or a shift in the reading frame.
About half of the variants in the protein-coding region of a genome are categorized as silent and expected to have no impact on the resulting protein structure. Among the remaining ~13,000 variants are several types of putative loss of function variants as well as missense variants.
Loss of function (LOF) variants are those that are predicted to disrupt the protein function in a significant way. These variants substantially truncate or entirely eliminate protein-coding transcripts and include stop-gained, stop-lost, frameshift insertion/deletion, and splice site disruptors. They are the most deleterious and best candidates for pathogenic variants. However, for some genes, it is not a loss of function but rather a gain of function that results in disease. Also, the location of protein-truncating variants is important. Those that occur mid-protein may have a greater impact relative to those that occur close to either end.
Missense variants can also be pathogenic, but it’s difficult to know if any given missense variant will impact the protein in such a way as to cause disease. The likelihood that a missense variant is deleterious (protein damaging) can be predicted based on features like conservation, amino acid substitution, and location using scores generated from algorithms like PhyloP, SIFT, and MutationTaster. Algorithms that combine individual predictors into a single measure, such as CADD or VVP8 (developed by Fabric), are sometimes even better at prioritizing variants based on the likelihood of damage.
Determining how common variants are in the population
Rare diseases are found in few individuals in the population. It follows that the disease-causing variants should be rare as well. Several public databases are the source of information about population allele frequencies. These include the ExAC, EVS, and 1K Genomes databases and the newer, more comprehensive gnomAD database. These databases aggregate genome/exome sequence data across thousands of individuals from different studies and are meant to represent an ostensibly healthy cohort in which variants causing rare genetic disease should be present at an extremely low frequency or not at all.
Confirming Mendelian inheritance of the disease
Parental genotypes are useful for confirming the mode of inheritance, as well as narrowing down the list of possible causal variants further. Trios (patient and both parents) are most commonly used, but the inclusion of other siblings, affected or unaffected, provides additional useful information as well.
For a classic autosomal dominant disorder, one parent would also be expected to have the disorder. A trio with both parents unaffected may represent a classic autosomal recessive or X-linked disorder or a de novo variant for a dominant disorder. The vast majority of genetic conditions are inherited autosomally, with slightly more recessive than dominant. De novo pathogenic variants are well described in about 15% of disease genes.9
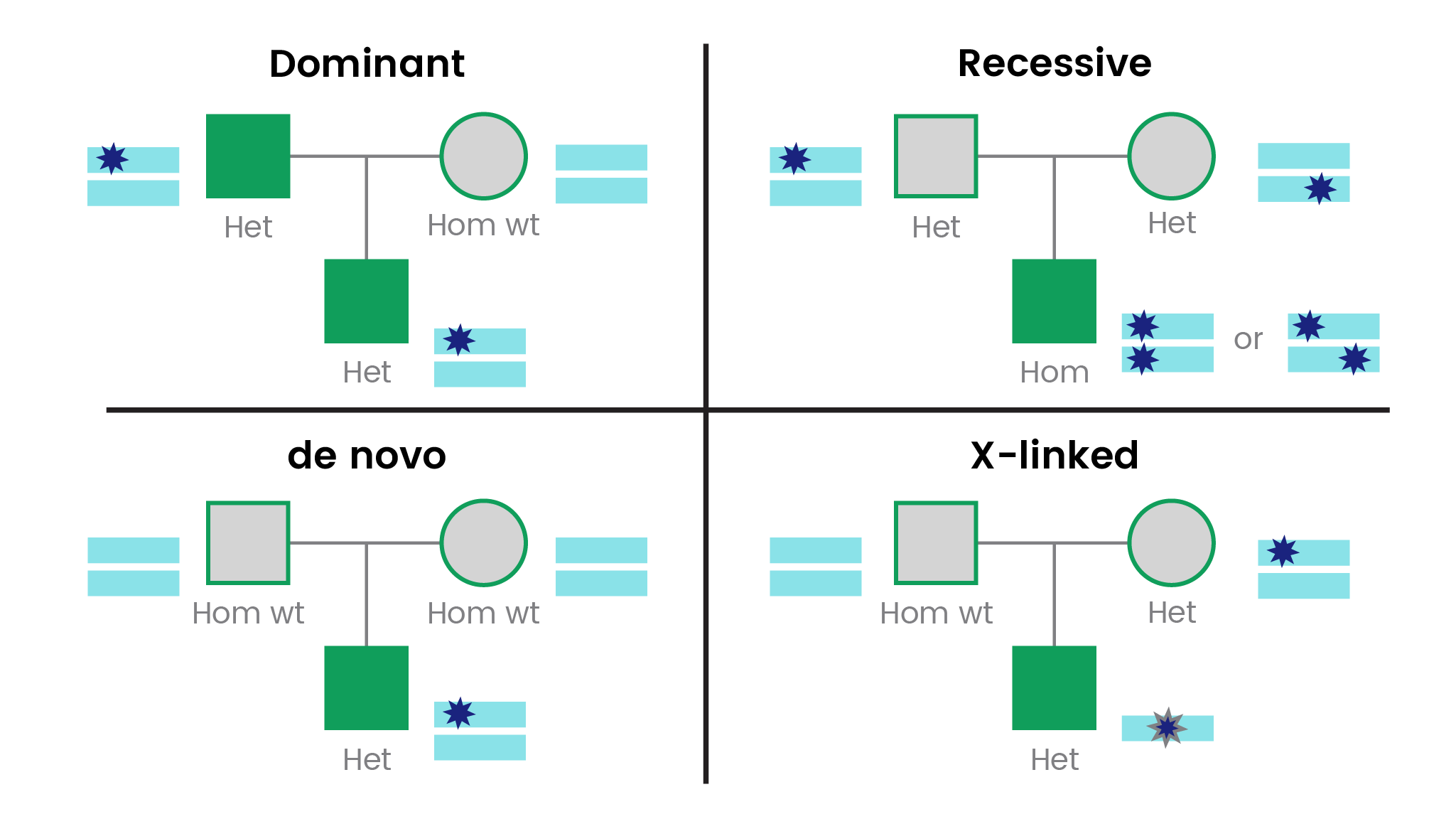
Autosomal recessive diseases due to the same mutation inherited from both parents are easy to identify as patients will be homozygous, and each parent heterozygous for the same variant. This type of autosomal recessive inheritance is more common in closed populations or in the context of consanguinity. Compound heterozygotes, where a different deleterious variant in the same gene is inherited from each carrier parent, is another equally likely scenario. In this case, parental genotypes can be used to confirm that the two deleterious variants in the patient are indeed affecting both copies of the gene (in trans) versus both being present in the same copy of the gene (in cis).
First Generation Genome Analysis – Variant Filtering

The earliest approaches used to identify causal variants from sequence data were based on variant filtering. Using the criteria discussed above – protein-altering, rare allele, Mendelian inheritance – one could easily go from an average of 4-5 million variants in a whole genome down to several hundred candidate variants.
While this represents a massive reduction in potential causal variants, it has some shortcomings. First, there are significant problems with applying hard cut-offs to the data when filtering. Depending on the cut-off, filtering can result in premature or inadvertent removal of causative candidates from the consideration set. At the same time, it may also generate large numbers of false-positive candidates. Second, filtering still requires several hundred genes to be evaluated, which is time-consuming and challenging to manage. It is estimated that each variant-gene assessment can take an expert about 15-30 minutes, potentially resulting in dozens of hours in review time. Filtering can be streamlined by working in a software environment that automates these processes and provides flexible tools to customize and track filters that are applied to the data. Fabric’s software includes these basic features to allow filtering and offers advanced algorithms to improve the efficiency of finding a causal variant dramatically.
Second Generation Genome Analysis – Gene Prioritization Algorithms
Filtering strategies alone are not a scalable solution for routine analysis of genomes for the clinical diagnosis of rare diseases. More sophisticated approaches are required to hone in on causal variants. Through a long-standing collaboration with Professor Mark Yandell at the University of Utah, Fabric has developed and deployed in its software the most cutting-edge disease gene identification algorithms.
VAAST – the first variant prioritization tool
In 2011, Fabric introduced the very first variant prioritization tool called VAAST (Variant Annotation, Analysis & Search Tool).10 VAAST is a gene-ranking algorithm that integrates information about phylogenetic conservation, amino acid substitution effects, allele frequencies, and other factors into a single unified likelihood-framework for ranking variants according to deleteriousness. This is particularly attractive for missense variants, the most common and most challenging variant type to analyze. VAAST scores each variant in a gene and uses a ‘burden test,’ which takes into account the effects of multiple variants in a gene simultaneously to produce a gene score. The gene score is particularly powerful for identifying recessive disorders caused by compound heterozygotes.
PHEVOR – integrating patient phenotypes into variant prioritization
Around this time, efforts to standardize the vocabulary around medically relevant phenotypes and their known association with genes resulted in the creation of the Human Phenotype Ontology (HPO). The HPO database provided a critical link from the patient’s phenotype to a list of candidate genes. Fabric developed its second variant prioritization algorithm called PHEVOR (the Phenotype Driven Variant Ontological Re-ranking tool) in 2014.11 PHEVOR builds upon the output of variant prioritization tools such as VAAST, integrating it with knowledge resident in multiple diverse biomedical ontologies. Ontology annotations, such as those found in Human Phenotype Ontology (HPO) and Gene Ontology (G,O), are readily available for many human and model organism genes. Starting with a list of phenotypes or gene function terms, PHEVOR leverages a network of these ontologies to automatically derive a candidate gene list from these terms, including genes that are not explicitly annotated to a given phenotype but for which the ontology graph structure implies potential latent linkages. It then re-ranks the variants from VAAST output accordingly, reprioritizing them in light of gene function, disease, and phenotype knowledge1.
Importantly, PHEVOR is not limited to known diseases or known disease-causing genes, making it suitable for research and clinical discoveries. PHEVOR also provides a means of integrating ontologies that contain knowledge not explicitly linked to phenotype (such as the GO) into the variant-prioritization process. Thus, PHEVOR can use information latent in such ontologies to discover disease-causing alleles in genes not previously associated with a disease or identify causative alleles in challenging cases with atypical symptoms.
GEM – improving the speed and efficiency of clinical diagnosis
In the last decade, thousands of patients have undergone clinical sequencing to diagnose their rare disease. Countless more have undergone sequencing in a research setting. These efforts are producing about 250 new Mendelian disease gene discoveries each year.9 Moreover, as routine genetic testing has ramped up, so has the cumulative body of knowledge around individuals’ variants and their relationship to disease. Information about disease genes and variants, cataloged in the OMIM and ClinVar databases, respectively, have become an indispensable resource for diagnosing rare diseases and laying the groundwork for treatment.
Fabric’s latest gene-ranking algorithm, GEM, was released in October 2020. GEM builds on VAAST and PHEVOR, but in addition, incorporates and prioritizes clinical information from OMIM and ClinVar. GEM infers ancestry to refine allele frequency cutoffs and uses probabilistic modeling to remove common sequencing artifacts. These Artificial Intelligence (AI) methods dramatically improve the speed, efficiency, and effectiveness of clinical genome interpretation. GEM takes as inputs the patient’s clinical features along with their genome sequence and produces a concise ranked list of clinically relevant variants. Benchmarking data across several cohorts of previously diagnosed rare genetic disease patients has shown the algorithm to be highly sensitive for finding known disease genes, with the causal gene ranking in the top 2 over 90% of the time. GEM consistently outperforms other algorithms in its ability to detect causal variants while reviewing the smallest number of genes.
An important feature of GEM is that it can analyze not only small variants (SNVs and short insertion and deletions) but also can discover and interpret structural variants. This feature enhances the ability to identify compound heterozygotes responsible for a recessive disease where one variant is an SV, resulting in an increased diagnostic yield. Gem also accounts for many common confounders that contribute to lengthy review times: variant call artifacts, regions of homozygosity due to consanguinity; patient ancestry; variable penetrance, and inheritance mode. The result is a concise list of strongly supported candidate genes that need review.
GEM’s succinct output is delivered through an intuitive user interface that streamlines the variant analysis workflow, allowing the clinician to focus on the most important data. Additionally, GEM is transparent, presenting component scores that support the rationale behind its variant prioritization.
GEM is positioned to dramatically increase the speed and efficiency of clinical genome analysis, with applications in the analysis of undiagnosed rare disease, rapid analysis of NICU/PICU cases and routine re-analysis of negative genomes.
Summary
Fabric Genomics has a long history of developing cutting-edge AI algorithms for analyzing genomes. Its latest algorithm, GEM, builds on over a decade of work and brings a first-in-class tool for clinical genome analysis of patients with rare genetic diseases. VAAST, PHEVOR, and GEM are integrated within the Fabric Enterprise software environment, which provides exhaustive annotations, standardized analysis workflows, and clinical reporting, providing an end-to-end solution for clinical labs to scale clinical analysis of genomes.
References
1. Worthey EA, Mayer AN, Syverson GD, et al. Making a definitive diagnosis: successful clinical application of whole exome sequencing in a child with intractable inflammatory bowel disease. Genet Med. 2011;13(3):255-262.
2. Clark MM, Stark Z, Farnaes L, et al. Meta-analysis of the diagnostic and clinical utility of genome and exome sequencing and chromosomal microarray in children with suspected genetic diseases. NPJ Genom Med. 2018;3:16.
3. Truty R, Paul J, Kennemer M, et al. Prevalence and properties of intragenic copy-number variation in Mendelian disease genes. Genet Med. 2019;21(1):114-123.
4. Stark Z, Tan TY, Chong B, et al. A prospective evaluation of whole-exome sequencing as a first-tier molecular test in infants with suspected monogenic disorders. Genet Med. 2016;18(11):1090-1096.
5. Tan TY, Dillon OJ, Stark Z, et al. Diagnostic Impact and Cost-effectiveness of Whole-Exome Sequencing for Ambulant Children With Suspected Monogenic Conditions. JAMA Pediatr. 2017;171(9):855-862.
6. van Nimwegen K, Vissers L, Willemsen M, et al. The Cost-Effectiveness of whole-Exome Sequencing in Complex Paediatric Neurology. Value in Health. 2016;19(7):A695.
7. Yeung A, Tan NB, Tan TY, et al. A cost-effectiveness analysis of genomic sequencing in a prospective versus historical cohort of complex pediatric patients. Genetics in Medicine. 2020.
8. Flygare S, Hernandez EJ, Phan L, et al. The VAAST Variant Prioritizer (VVP): ultrafast, easy to use whole genome variant prioritization tool. BMC Bioinformatics. 2018;19(1):57.
9. Bamshad MJ, Nickerson DA, Chong JX. Mendelian Gene Discovery: Fast and Furious with No End in Sight. Am J Hum Genet. 2019;105(3):448-455.
10. Hu H, Huff CD, Moore B, Flygare S, Reese MG, Yandell M. VAAST 2.0: improved variant classification and disease-gene identification using a conservation-controlled amino acid substitution matrix. Genet Epidemiol. 2013;37(6):622-634.
11. Singleton MV, Guthery SL, Voelkerding KV, et al. Phevor combines multiple biomedical ontologies for accurate identification of disease-causing alleles in single individuals and small nuclear families. Am J Hum Genet. 2014;94(4):599-610.