

BLOG
Avoiding the Common Mistakes Labs Make When Launching Hereditary Cancer Panel Tests
By Jeanette McCarthy, MPH, PhD, Sr. Director Scientific Programs
Introduction
The Current Hereditary Cancer Testing Landscape
Hereditary cancer tests are among the most common genetic tests available, with over 600 testing products on the market. These tests cover the analysis of single nucleotide variants (SNVs) as well as copy number variants (CNVs) and range in size from individual genes to panels of up to hundreds of genes. Hereditary cancer gene panels can be built around a single cancer, groups of related cancers, or even all known hereditary cancers. There are currently over 100 pan-cancer panels on the market and many cancer-specific panels, especially for breast/ovarian and colorectal cancers. These tests are being utilized among cancer patients to determine their prognosis or risk of developing other cancers and for determining treatment response. For example, response to PARP inhibitors is related to the presence of inherited variants in the BRCA genes. Testing is also being used in ostensibly healthy people, especially those with a family history of cancer, who want to know their risk of developing cancer.
Clinical Utility, Guidelines, and Reimbursement
Hereditary cancer testing is gaining acceptance in the medical community. Testing recommendations have been incorporated into the professional guidelines of over six organizations, including the National Comprehensive Cancer Network. Although it is quite nuanced, insurance reimbursement for these tests has also improved. For example, Medicare provides coverage for hereditary cancer testing in patients with a personal history of breast, ovarian, colorectal, and other cancer types. Private insurance is following suit, although there is quite a bit of variability in precisely who they cover when the testing is done and which tests are used. The Affordable Care Act requires insurance companies to pay for both genetic counseling and HBOC testing in women who meet specific criteria. This broad acceptance among professional societies coupled with improving reimbursement landscape explains why these tests appear on the menus of so many diagnostic labs.
Professional Guidelines |
|---|

|
insurance coverage |
|---|
Medicare coverage: Hereditary breast and ovarian cancer (HBOC) and Lynch testing in patients with a personal history of cancer
Private insurance: Most policies written very narrowly – specific patients, timing, tests Affordable Care Act: Insurance companies are required to pay for both genetic counseling and BRCA testing for women who meet certain criteria |
Requirements for Launching Panels
To launch a hereditary cancer test in a lab, there are several things to consider:
- The design of your gene panel – this is where you select the genes that will go on the panel, with consideration to both the clinical validity and utility, customer demand, as well as reimbursement.
- The wet lab component involves selecting an assay and sequencing method and implementing this process in a CLIA-regulated environment.
- Informatics capabilities are essential, including a means of calling, annotating, visualizing, and classifying variants
- Having the right personnel for interpreting genetic variants, creating and signing out clinical reports.
- There is also an overarching need for a robust validation process. Perhaps the biggest mistake labs make is underestimating what is required for validation of NGS-based panel tests. Labs with experience launching other types of tests may not appreciate the nuances involved, and their process can quickly get derailed. For instance, labs sometimes do not choose the right validation samples to match the variants that the test is intended to cover. This can cause significant delays in launching a test.
Common Mistakes When Launching Panel Tests
Common Mistake 1: Not weighing the pros and cons of including genes with limited clinical validity
There is a common misconception that because an off-the-shelf assay is available for a very large number of genes, you should automatically include all of them on your panel. Assays like the Illumina Trusight 113 gene hereditary cancer panel can be run as a backbone from which several smaller virtual panels can be offered. However, this panel contains a mix of well-vetted, clinically valid genes along with more investigational genes without strong evidence of association with cancer. Simply reporting out on all of the genes in this assay can be problematic for several reasons.
First, in some cases, insurance companies may only cover gene panels where ALL genes in the panel have demonstrated clinically valid and utility.
Second, while some genes are associated with multiple cancer types, the level of evidence for association with each individual cancer type needs to be considered. Take, for example, the five genes associated with Lynch syndrome, where definitive evidence of association and risk for colorectal cancer has been determined, but where the association with breast cancer is tenuous. Inclusion of these genes on hereditary breast cancer panels is problematic when testing healthy women where data does not exist to tell them their risk of breast cancer. The point is that careful consideration needs to be given to the individual genes and their intended use.
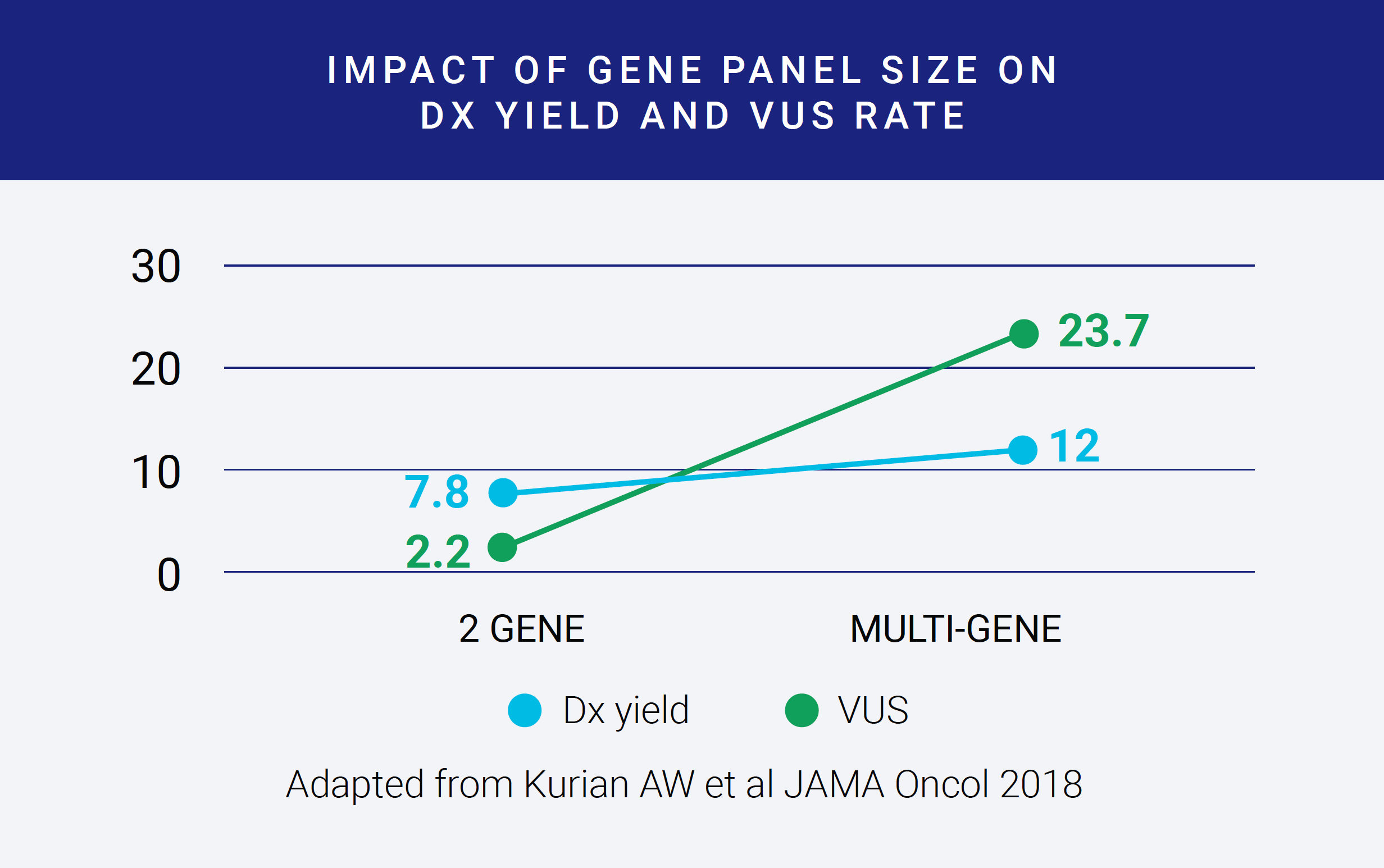
The final reason to restrict a gene panel’s size is that larger panels that include less well-validated genes can result in increased rates of variants of uncertain significance. In this figure, adapted from a paper in JAMA Oncology, the diagnostic yield of the test, shown as the blue line, is improved modestly when going from a two-gene to a multi-gene panel. In contrast, the VUS rate, shown as an orange line, increases dramatically. The VUS rates reported here are for Whites. Rates in Blacks and Asians are much, much higher. More VUS’s require more interpretation time and thus can become costly, and for possibly minimal improvement in diagnostic yield. Having a solution for streamlining variant interpretation is vital for controlling costs for larger panels.
Common Mistake 2: Selecting an assay before considering genes, variants, and future testing needs
For most genes included on hereditary cancer panels, NGS alone may be sufficient to yield high sensitivity and specificity. However, there are some well-known gaps in sequence coverage in key genes. For this reason, target enrichment assays are often utilized to reduce these gaps and improve sensitivity. Different assays employ distinct enrichment strategies and baits, leading to variability in the quality of downstream sequencing data. Some assays provide a more uniform breadth of coverage than others, leading to more accurate variant identification.
One of the biggest mistakes labs make is selecting an assay before considering which specific genes and variants need to be queried, how well a specific assay behaves for those genes and types of variants, or considering how their testing menu may expand in the future. Spending time considering these aspects prior to selecting an assay can save a lot of headaches later on.
Labs may choose to utilize large off-the-shelf assays or to develop a smaller custom assay. Smaller assays may be more economical but can limit downstream use of the panel for reflex testing. For example, you may only be interested in offering a breast cancer panel today, but you may want to expand to offer a pan-cancer panel a year from now. Similarly, you may only be interested in including clinically valid genes on your panel today but choosing a narrow assay may prohibit you from offering reflex testing with more investigational genes later.
It is also essential to have a sense of how critical CNVs are for all of the genes on your panel and whether the assay is suitable for detecting those. If not, orthogonal methods should be used to detect these CNVs. You should also understand what regions of the gene are likely to contain pathogenic variants. For example, are pathogenic variants ever found in the UTR or intronic regions? If so, you’ll need to ensure adequate coverage of those regions in the assay.
| Breadth of coverage | For higher confidence variant calling |
| # of genes covered | Balance cost and flexibility |
| Variation detected | SNVs, CNVs? |
| Exons; UTRs, full or partial introns | Target regions |
Problem: picking off the shelf assay before considering what genes/variants are required for your panel test.
Finally, determining the appropriate depth of sequencing coverage that will be required to detect specific types of variants in different gene regions is critical, as is considering which quality control metrics will be used to ensure the analytical validity of your test.
After sequence data are generated, and the variants identified, the process moves into the realm of data analytics, where having a robust software solution can dramatically improve the accuracy, consistency, efficiency, and cost of delivering gene panel results at scale.
Common Mistake 3: Not applying variant classification criteria consistently
Sequencing a gene panel will likely identify variants in everyone, but not all of those variants are pathogenic or disease-causing. To classify a variant as pathogenic or benign, multiple lines of evidence need to be considered simultaneously – phenotype, databases, prior cases, computational predictors, and literature. Each laboratory develops its own variant classification method, typically based on ACMG/AMP guidelines. ACMG guidelines include 28 criteria that are combined using a set of rules to achieve a final classification of either pathogenic, likely pathogenic, benign, likely benign, or a variant of uncertain significance. For labs offering gene panel tests, the interpretation component can add significant time and cost as it requires highly trained staff, is sometimes done manually, and is time-consuming.
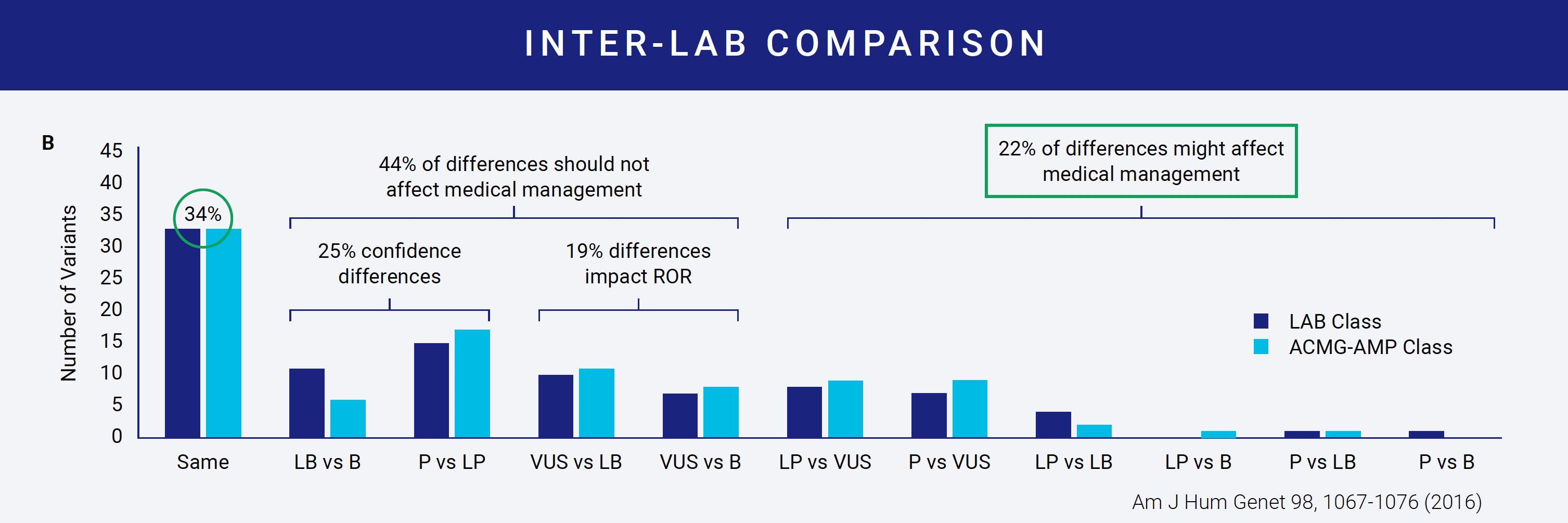
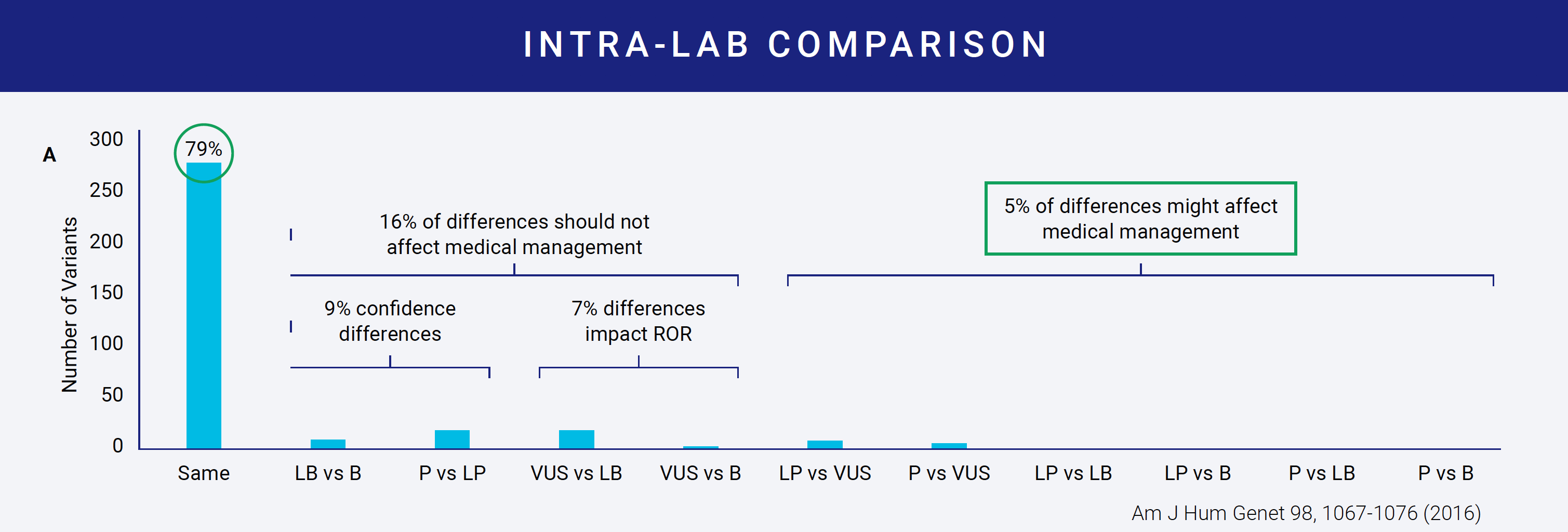
Despite available interpretation guidelines, the same variant can be classified differently by two labs. In one study of nine labs interpreting the same variants, concordance of variant classification between labs was achieved only 34% of the time. Even more interesting was that within a lab, concordance only reached 79%.
Reasons for discordant interpretations become evident when you examine even the most straightforward criteria. For example, several criteria ask about the variant allele frequency (AF). There are four public databases that most, if not all, labs use to get AF information. The table on the right shows how the AF for a single variant across the four databases can vary. Which AF do you use? The lowest? The highest? Does it need to be consistently high or low across all? Do you use the overall database frequency or examine continental subpopulation frequencies? The range here is even greater. Depending on what you decide, your answers to the criteria can vary and impact the downstream classification. This is why most labs develop protocols for variant interpretation that the different variant scientists can follow. These protocols define how each of the criteria should be answered and how each of the rules should be combined.
Hardcoding these protocols in a flexible software application saves time and also improves the consistency of interpretation between reviewers, ensuring a high level of precision.

Common Mistake 4: Not leveraging prior information during variant interpretation to improve efficiency
Variant interpretation can be a labor-intensive process and contributes significantly to the overall cost of delivering a gene panel test. Any efforts to the efficiency of the variant interpretation process can reduce the underlying cost of a panel test. Efficiencies can be gained in two areas. The first is around re-using gene-specific knowledge, and the second is around re-using variant classifications.
Gene-specific knowledge feeds into many of the ACMG criteria. Examples of relatively stable knowledge about a gene include the mode of inheritance, penetrance, age of onset, and prevalence of the gene’s disorder. For well-established disease genes, additional knowledge becomes stable over time, such as the rate of benign and rate of pathogenic missense variation in the gene or whether the mechanism of disease is loss of function or gain of function.
Having this gene-specific information available upfront to variant interpreters at the point of interpretation means two things. First, there is no need for the variant scientist to spend time gathering critical information about the gene every time they see a variant in the gene. Second, if two different variant scientists use the same underlying gene information, it will improve the consistency of interpretations. Therefore, software tools that not only display but also integrate pre-curated gene information into the interpretation platform can save time and also improve consistency within a lab.
At Fabric, we offer pre-curated panels for customers who use our software. Our curation scientists curate each panel gene upfront using a wide range of data sources. This codified gene curation data feeds into our automated variant classification engine, ACE, and is also visible during manual variant interpretation.
Efficiencies in interpretation can also be gained at the level of the variant. At Fabric, we have developed a classified variant database. This database gives labs a place to store any variant that they classify, along with variant descriptions, and then the ability to quickly call up those variants if they are seen again. This database allows a user to save and share classified variants internally with the option to share and pull knowledge from other sites. There are different levels of sharing to protect patient privacy.
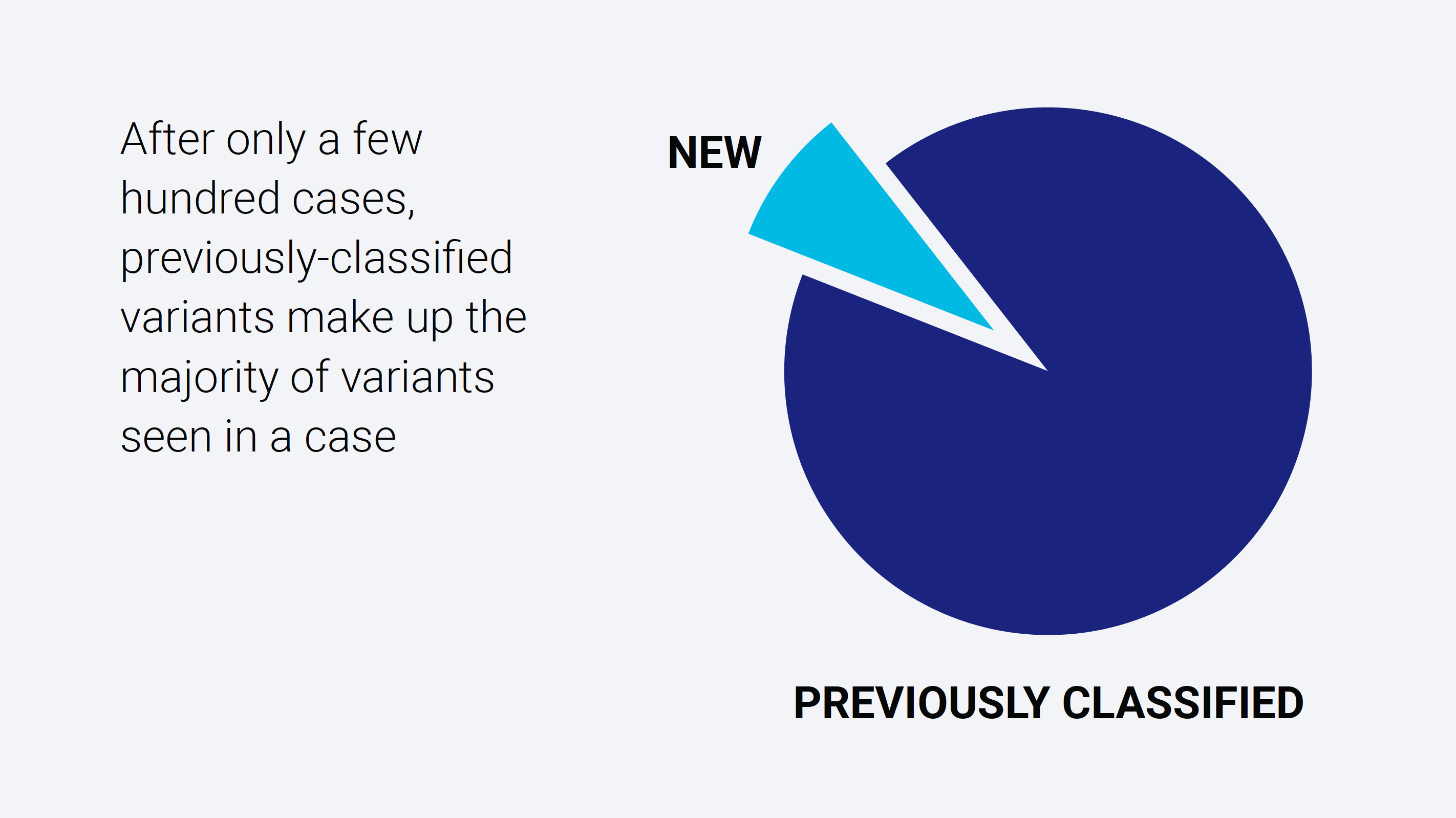
After only a few hundred cases, the classified variant database becomes populated to a point where the effective number of variants requiring review drops dramatically.
Summary
Hereditary cancer panels are becoming increasingly important clinically, and their use is growing quickly. As more labs expand their offering to include these tests, it is essential to have a well-designed process that avoids the common mistakes and is accurate, scalable, and profitable from the start. Fabric genomics offers an end-to-end solution to help labs launch hereditary cancer panels quickly. Leveraging software such as Fabric Enterprise, Fabric Ace, and the associated pre-curated panels and classified variant database will maximize accuracy, consistency, and efficiency from the start. We also offer comprehensive services to design, validate and launch panel testing, as well as a full suite of clinical services to assist in everything from interpretation to sign out.
SUBSCRIBE